
This post is about the following paper (and the figures are reproduced from it and are copyright of the American Physical Society): “Machine learning force fields based on local parametrization of dispersion interactions: Application to the phase diagram of C60“, by Heikki Muhli, Xi Chen, Albert P. Bartók, Patricia Hernández-León, Gábor Csányi, Tapio Ala-Nissila, and Miguel A. Caro. Phys. Rev. B 104, 054106 (2021). A preprint is openly available from the arXiv.
Adding dispersion (or van der Waals [vdW]) corrections to density functional theory (DFT) has been a very active area of research in the past 10-15 years. DFT is a mean field theory and dynamical effects, such as the effects of fluctuating charge distributions on energy and forces, are notoriously missing. The leading term in these vdW corrections is the London dispersion force, which decays as the sixth power of the interatomic distance (Vij ∝ C6 / rij6). While this power law indicates that these interactions quickly become very small with distance for any given two atoms, they are usually attractive. This means that the collective effect of vdW interactions can lead to interesting emerging physical phenomena. An important example of this is how individual graphene layers interact with one another to form graphite.
The first vdW correction scheme to achieve widespread adoption was the D2 method, due to Stefan Grimme (Grimme, J. Comp. Chem. 27, 1787 [2006]). In this method, the effective C6 coefficient that parametrizes the London dispersion formula is given as a function of the atomic species and a damping function was introduced to switch off the dispersion correction at short interatomic distances. Soon, more sophisticated approaches arose that improved the accuracy of vdW corrections, by incorporating both 1) more information about the dependence of the effective C6 coefficient on the local atomic environment and 2) improved damping functions that bridge the transition between the long-range (London-type) correlation energy and the short-range DFT correlation energy. The Tkatchenko-Scheffler correction scheme (Tkatchenko and Scheffler, Phys. Rev. Lett. 102, 073005 [2009]) is one such approach, which relies on an “atom in a molecule” approximation and relates the effective C6 coefficients and damping length scales to the Hirshfeld partitioning of the charge density.
In parallel to these efforts on dispersion-correction schemes, the community has made huge advances in the past 10 years on machine learning interatomic interactions, usually from DFT data. These machine learning (ML) force fields are normally based on a local (atom-wise) decomposition of the total energy, to keep the simulation problem tractable. This is fine for strong covalent and repulsion interactions, which are typically short ranged. For instance, in carbon materials the covalent (“bonded”) part of the total energy of an atomistic system can be accurately learned with local atomic descriptors that are “blind” beyond 4-5 Angstrom. But vdW interactions are long ranged and, depending on the level of detail one aims at capturing, the “local” atomic environment relevant to vdW interactions is of the order of 15-20 Angstrom. This is bad news, because the computational cost of ML force fields scales as the cube of this distance, known as “cutoff”. This means that a ML force field with a 20 Angstrom cutoff is approximated 64 times as expensive, computationally, as another ML force field with a 5 Angstrom cutoff. Less obvious, but equally severe, limitations of “brute forcing” the learning of long-range interactions include the explosion of the size of configuration space with the cutoff, which requires exponentially more data for training these models.
Making ML potentials two orders of magnitude slower is an unacceptable tradeoff for including vdW corrections. The approach we, and also others before us, took in our recent paper is to machine learn the Hirshfeld volumes, which are a function of the local atomic environment with locality similar to the covalent part of the total energy. Then, these Hirshfeld volumes are used to parametrize the Tkatchenko-Scheffler implementation of the London dispersion equation, which is computationally cheap to evaluate. In addition, we took care of efficiently coupling the covalent and vdW parts of the calculation, such that the overhead due to the Hirshfeld volume computation is very small, and the overall vdW-enabled force field is, for typical calculations, only between 20% and 50% more expensive than the force field without vdW corrections. A happy consequence of our ML implementation is that forces can be computed more accurately than the reference method, because the gradients of the Hirshfeld volumes can be computed generally with ML, whereas the DFT implementation cannot easily compute these terms (which are typically missing from DFT calculations). We implemented these corrections in the GAP and TurboGAP codes.
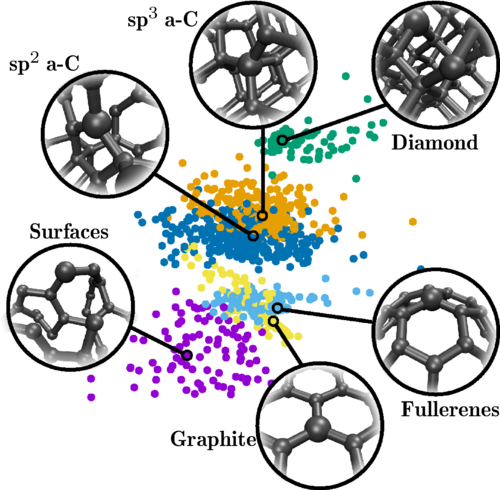
As a proof of concept, we trained a new GAP for carbon and fine tuned it specifically to simulate C60, a carbon material entirely made of C60 molecules. We could simulate large systems with thousands of atoms for relatively long MD times, and could chart the metastable high temperature/high pressure phase diagram of C60, observing the transformations to graphitic carbon and amorphous carbon taking place in this material under specific thermodynamic conditions (see feature image at the top of this post).
This work is the culmination of a long effort by our group’s PhD student Heikki Muhli, that started more than 2 years ago during his MSc thesis project and continued as part of our COMPEX project, funded by the Academy of Finland, with CPU time provided by CSC and Aalto University’s Science IT project. It is also part of our continuous effort to develop and improve the TurboGAP code, which we hope to be able to launch within the next few months (the code’s development version is available online for the brave). For this work we collaborated with our group’s other PhD student Patricia Hernández-León, Aalto Univerty’s Xi Chen and Tapio Ala-Nissila, and GAP authors Albert Bartók (Warwick) and Gábor Csányi (Cambridge).
And this is just the beginning. Newer vdW correction schemes worry about the role of “many-body” physics on vdW corrections, where the leading effect is how many-body interactions affect the effective C6 coefficients (Otero-de-la-Roza, LeBlanc and Johnson, Phys. Chem. Chem. Phys. 22, 8266 [2020]). One of these many-body dispersion methods is the so-called “many-body dispersion” method (MBD; early bird gets the worm), which also feeds on Hirshfeld charge density partitioning (Tkatchenko, DiStasio Jr., Car and Scheffler. Phys. Rev. Lett. 108, 236402 [2012]). With our new infrastructure to couple GAPs to simpler, locally parametrized, force fields, we will be able in the near future to incorporate many-body dispersion effects, as well as other long-range interactions, such as electrostatics.

